For decades, researchers worked together to assemble a complete copy of the molecular instructions for a human — a map of the human genome. The first draft was finished in 2000, but with several missing pieces. Even when a complete reference genome was achieved in 2022, their work was not finished. A single reference genome can’t incorporate known genetic variations, such as the variants for the gene determining whether a person has a blood type A, B, AB or O. Furthermore, the reference genome didn’t represent the vast diversity of human ancestries, making it less useful for detecting disease or finding cures for people from some backgrounds than others. For the past three years, we have been part of an international collaboration with 119 scientists across 60 institutions, called the Human Pangenome Research Consortium, to address these challenges by creating a new and more representative map of the human genome, a pangenome.
We are excited to share that today, in “A draft human pangenome reference”, published in Nature, this group is announcing the completion of the first human pangenome reference. The pangenome combines 47 individual genome reference sequences and better represents the genomic diversity of global populations. Building on Google’s deep learning technologies and past advances in genomics, we used tools based on convolutional neural networks (CNNs) and transformers to tackle the challenges of building accurate pangenome sequences and using them for genome analysis. These contributions helped the consortium build an information-rich resource for geneticists, researchers and clinicians around the world.
 |
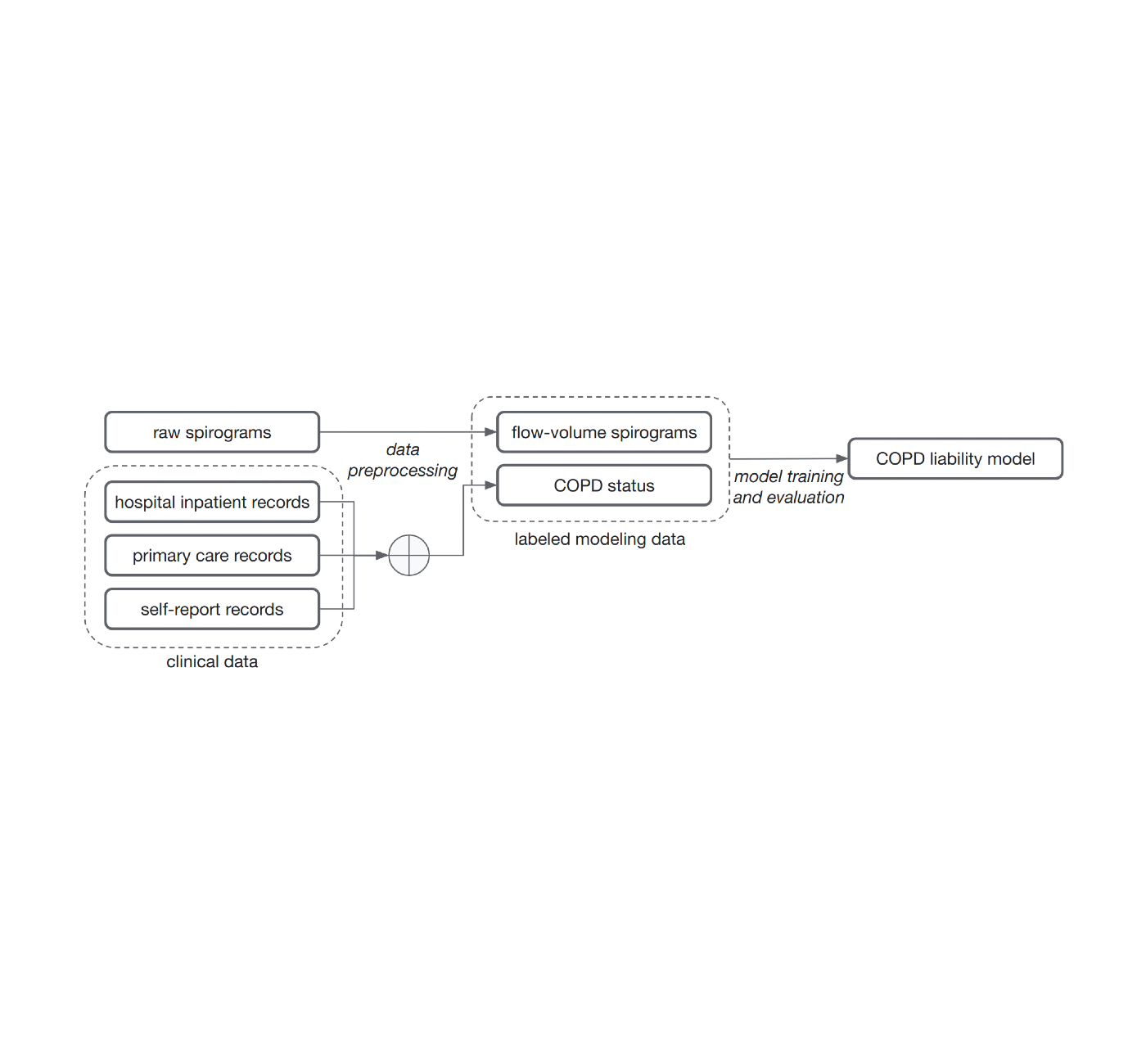
Using graphs to build pangenomes
In the typical analysis workflow for high-throughput DNA sequencing, a sequencing instrument reads millions of short pieces of an individual’s genome, and a program called a mapper or aligner then estimates where those pieces best fit relative to the single, linear human reference sequence. Next, variant caller software identifies the unique parts of the individual’s sequence relative to the reference.
But because humans carry a diverse set of sequences, sections that are present in an individual’s DNA but are not in the reference genome can’t be analyzed. One study of 910 African individuals found that a total of 300 million DNA base pairs — 10% of the roughly three billion base pair reference genome — are not present in the previous linear reference but occur in at least one of the 910 individuals.
To address this issue, the consortium used graph data structures, which are powerful for genomics because they can represent the sequences of many people simultaneously, which is needed to create a pangenome. Nodes in a graph genome contain the known set of sequences in a population, and paths through those nodes compactly describe the unique sequences of an individual’s DNA.
 |
| Schematic of a graph genome. Each color represents the sequence path of a different individual. Multiple paths passing through the same node indicate multiple individuals share that sequence, but some paths also show a single nucleotide variant (SNV), insertions, or deletions. Illustration credit Darryl Leja, National Human Genome Research Institute (NHGRI). |
 |
| Actual graph genome for the major histocompatibility complex (MHC) region of the genome. Genes in MHC regions are essential to immune function and are associated with a person’s resistance and susceptibility to infectious disease and autoimmune disorders (e.g., ankylosing spondylitis and lupus). The graph shows the linear human genome reference (green) and different individual person’s sequence (gray). |
Using graphs creates numerous challenges. They require reference sequences to be highly accurate and the development of new methods that can use their data structure as an input. However, new sequencing technologies (such as consensus sequencing and phased assembly methods) have driven exciting progress towards solving these problems.
Long-read sequencing technology, which reads larger pieces of the genome (10,000 to millions of DNA characters long) at a time, are essential to the creation of high quality reference sequences because larger pieces can be stitched together into assembled genomes more easily than the short pieces read out by earlier technologies. Short read sequencing reads pieces of the genome that are only 100 to 300 DNA characters long, but has been the highly scalable basis for high-throughput sequencing methods developed in the 2000s. Though long-read sequencing is newer and has advantages for reference genome creation, many informatics methods for short reads hadn’t been developed for long read technologies.
Evolving DeepVariant for error correction
Google initially developed DeepVariant, an open-source CNN variant caller framework that analyzes the short-read sequencing evidence of local regions of the genome. However, we were able to re-train DeepVariant to yield accurate analysis of Pacific Bioscience’s long-read data.
 |
| Training and evaluation schematic for DeepVariant. |
We next teamed up with researchers at the University of California, Santa Cruz (UCSC) Genomics Institute to participate in a United States Food and Drug Administration competition for another long-read sequencing technology from Oxford Nanopore. Together, we won the award for highest accuracy in the nanopore category, with a single nucleotide variants (SNVs) accuracy that matched short-read sequencing. This work has been used to detect and treat genetic diseases in critically ill newborns. The use of DeepVariant on long-read technologies provided the foundation for the consortium’s use of DeepVariant for error correction of pangenomes.
DeepVariant’s ability to use multiple long-read sequencing modalities proved useful for error correction in the Telomere-to-Telomere (T2T) Consortium’s effort that generated the first complete assembly of a human genome. Completing this first genome set the stage to build the multiple reference genomes required for pangenomes, and T2T was already working closely with the Human Pangenome Project (with many shared members) to scale those practices.
With a set of high-quality human reference genomes on the horizon, developing methods that could use those assemblies grew in importance. We worked to adapt DeepVariant to use the pangenome developed by the consortium. In partnership with UCSC, we built an end-to-end analysis workflow for graph-based variant detection, and demonstrated improved accuracy across several thousand samples. The use of the pangenome allows many previously missed variants to be correctly identified.
 |
| Visualization of variant calls in the KCNE1 gene (a gene with variants associated with cardiac arrhythmias and sudden death) using a pangenome reference versus the prior linear reference. Each dot represents a variant call that is either correct (blue dot), incorrect (green dot) — when a variant is identified but is not really there —or a missed variant call (red dot). The top box shows variant calls made by DeepVariant using the pangenome reference while the bottom shows variant calls made by using the linear reference. Figure adapted from A Draft Human Pangenome Reference. |
Improving pangenome sequences using transformers
Just as new sequencing technologies enabled new pangenome approaches, new informatics technologies enabled improvements for sequencing methods. Google adapted transformer architectures from analysis of human language to genome sequences to develop DeepConsensus. A key enabler for this was the development of a differentiable loss function that could handle the insertions and deletions common in sequencing data. This enabled us to have high accuracy without needing a decoder, allowing the speed required to keep up with terabytes of sequencer output.
 |
| Transformer architecture for DeepConsensus. DeepConsensus takes as input the repeated sequence of the DNA molecule, measured from fluorescent light detected by the addition of each base. DeepConsensus also uses as input the more detailed information about the sequencing process, including the duration of the light pulse (referred to here as pulse width or PW), the time between pulses (IP) the signal-to-noise ratio (SN) and which side of the double helix is being measured (strand). |
 |
| Effect of alignment loss function in training evaluation of model output. Better accounting of insertions and deletions by a differentiable alignment function enables the model training process to better estimate errors. |
DeepConsensus improves the yield and accuracy of instrument data. Because PacBio sequencing provides the primary sequence information for the 47 genome assemblies, we could apply DeepConsensus to improve those assemblies. With application of DeepConsensus, consortium members built a genome assembler that was able to reach 99.9997% assembly base-level accuracies.
Conclusion
We developed multiple new approaches to improve genetic sequencing methods, which we then used to construct pangenome references that enable more robust genome analysis.
But this is just the beginning of the story. In the next stage, a larger, worldwide group of scientists and clinicians will use this pangenome reference to study genetic diseases and make new drugs. And future pangenomes will represent even more individuals, realizing a vision summarized this way in a recent Nature story: “Every base, everywhere, all at once.” Read our post on the Keyword Blog to learn more about the human pangenome reference announcement.
Acknowledgements
Many people were involved in creating the pangenome reference, including 119 authors across 60 organizations, with the Human Pangenome Reference Consortium. This blog post highlights Google’s contributions to the broader work. We thank the research groups at UCSC Genomics Institute (GI) under Professors Benedict Paten and Karen Miga, genome polishing efforts of Arang Rhie at National Institute of Health (NIH), Genome Assembly and Polishing of Adam Phillipy’s group, and the standards group at National Institute of Standards and Technology (NIST) of Justin Zook. We thank Google contributors: Pi-Chuan Chang, Maria Nattestad, Daniel Cook, Alexey Kolesnikov, Anastaysia Belyaeva, and Gunjan Baid. We thank Lizzie Dorfman, Elise Kleeman, Erika Hayden, Cory McLean, Shravya Shetty, Greg Corrado, Katherine Chou, and Yossi Matias for their support, coordination, and leadership. Last but not least, thanks to the research participants that provided their DNA to help build the pangenome resource.