Posted by Rory Sayres PhD and Jonathan Krause PhD, Google AI, HealthcareTwo years ago, we announced
our inaugural work in training deep learning models for diabetic retinopathy (DR), a complication of diabetes that is one of the fasting growing causes of vision loss. Based on this research, we set out to
apply our technology to improve health outcomes in the world. At the same time, we’ve continued our efforts to improve the model’s performance, explainability, and applicability in clinical settings. Today, we are sharing our research progress toward these goals, as well as announcing a new partner in Thailand.
Improving Model Performance with High-quality LabelsThe performance of DR deep learning models is critically important, especially when subtle errors have the potential to generate a misdiagnosis. Earlier this year we published
a paper in the journal
Ophthalmology that looked at how we could improve our model by 1) moving toward a more granular 5-point grading scale (versus the previous 2-class system) and 2) incorporating adjudication by a panel of retinal specialists. During the adjudication process, a group of retinal specialists debated any case with disagreement until everyone agreed on the final grade. Compared to simply taking a majority vote, this method of resolving disagreements was more accurate and allowed for the identification of subtle findings, such as
microaneurysms.
To increase the efficiency of the adjudication process, we carefully selected a small subset (0.22%) of images to use as a tuning set, substantially improving model performance by optimizing model hyperparameters on this more accurate reference standard. When we subsequently measured the rate of agreement against a test set of images with an adjudicated reference standard, the kappa scores (a measurement of agreement that ranges from 0 [random] to 1 [perfect agreement]) for individual retinal specialists, ophthalmologists, and the algorithm ranged from 0.82-0.91, 0.80-0.84, and 0.84, respectively.
Making our Models More TransparentAs we deploy this technology, it is important that we take the proper steps to ensure that it is transparent and trusted. To that end, we have been exploring ways to explain how the model is making its predictions, with the goal of making the DR model a better diagnostic tool and aid for doctors.
In our latest study, to be published today in
Ophthalmology, we demonstrate methods by which explanations of deep learning algorithms can be shown to ophthalmologists to increase both the accuracy and confidence of their grading for diabetic eye disease. Using the results of the model trained and validated on high quality labels from our earlier study, we generated different forms of potential assistance for general ophthalmologists. We presented to the physicians the algorithm’s predicted scores for different DR severity levels as well as heatmaps highlighting image regions that most strongly drove its predictions. Using this assistance, we saw a significant increase in physicians’ diagnostic accuracy, as well as improved confidence in their diagnosis.
We saw clear evidence that showing model predictions could help physicians catch pathology they otherwise might have missed. In the retinal image below, our adjudication panel found signs of vision-threatening DR. This was missed by 2 of 3 doctors who graded it without assistance; but caught by all 3 doctors who graded it when they saw the model predictions (which accurately detected the pathology).
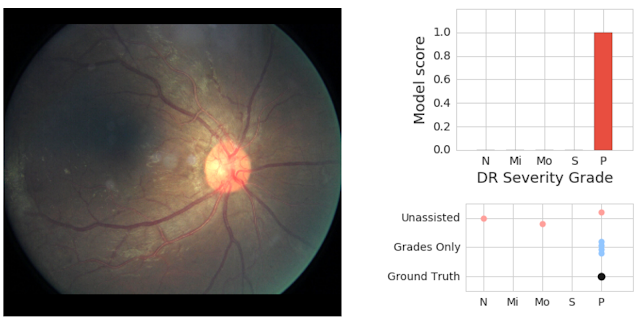 |
| On the left is a fundus image graded as having proliferative (vision-threatening) DR by an adjudication panel of ophthalmologists (ground truth). On the top right is an illustration of our deep learning model’s predicted scores (“P” = proliferative, the most severe form of DR). On the bottom right is the set of grades given by physicians without assistance (“Unassisted”) and those who saw the model’s predictions (“Grades Only”). |
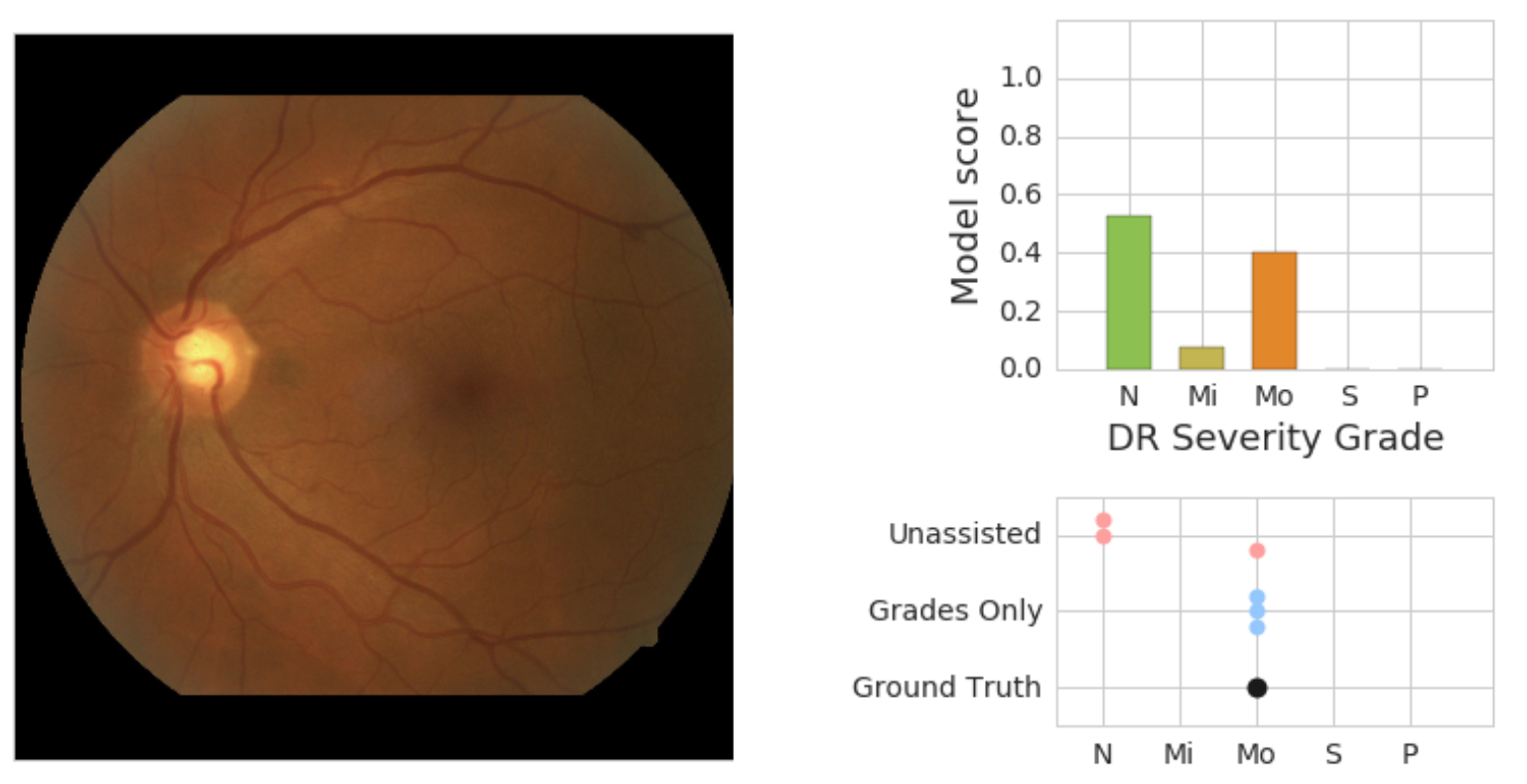
We also saw evidence that physicians and the model can work together in a way that provides more accuracy than either individually. In the retinal image below, our adjudication panel of retina specialists considered it to have moderate DR. Without assistance, two out of three ophthalmologists grading the image marked it as no DR. In real-world settings, this situation could result in a patient missing a needed referral to a specialist.
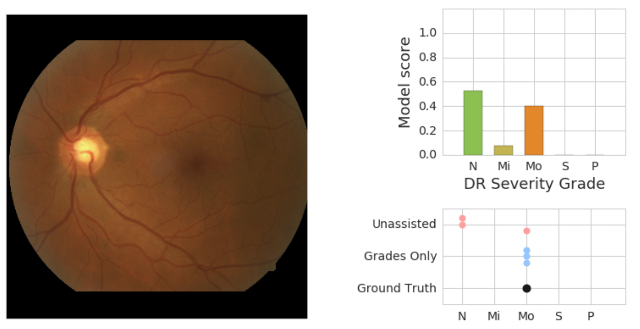 |
| On the left is a retinal fundus image graded as having moderate DR (“Mo”) by an adjudication panel of ophthalmologists (ground truth). On the top right is an illustration of the predicted scores (“N” = no DR, “Mi” = Mild DR, “Mo” = Moderate DR) from the model. On the bottom right is the set of scores given by physicians without assistance (“Unassisted”) and those who saw the model’s predictions (“Grades Only”). |
In this particular case, our model also indicated evidence for no DR. However, when ophthalmologists saw the model’s predictions, all three gave the correct answer. Seeing that the model saw some evidence for Moderate -- even if it wasn’t the highest score -- may prompt doctors to examine particular cases more carefully for pathology they may otherwise miss. We are excited to develop assistance that works like this, where human and machine learning abilities complement each other.
A New Partner in our Global EffortsWith the help of screening programs and in collaboration with
Verily, we have laid a robust foundation for the implementation of these highly accurate systems in real world clinical settings. Working with doctors at Aravind Eye Hospitals and Sankara Nethralaya in India, and now, through our new partnership with the
Rajavithi Hospital, affiliated with the Department of Medical Services, Ministry of Public Health in Thailand, we are validating the model performance with patients from broad screening programs. Given the positive results of our model on their real patient population, we are now beginning to pilot the model in their screening programs. We’re looking forward to a very busy 2019!