Posted by Ameya Velingker, Research Scientist, Google Research, and Balaji Venkatachalam, Software Engineer, Google

Graphs, in which objects and their relations are represented as nodes (or vertices) and edges (or links) between pairs of nodes, are ubiquitous in computing and machine learning (ML). For example, social networks, road networks, and molecular structure and interactions are all domains in which underlying datasets have a natural graph structure. ML can be used to learn the properties of nodes, edges, or entire graphs.
A common approach to learning on graphs are graph neural networks (GNNs), which operate on graph data by applying an optimizable transformation on node, edge, and global attributes. The most typical class of GNNs operates via a message-passing framework, whereby each layer aggregates the representation of a node with those of its immediate neighbors.
Recently, graph transformer models have emerged as a popular alternative to message-passing GNNs. These models build on the success of Transformer architectures in natural language processing (NLP), adapting them to graph-structured data. The attention mechanism in graph transformers can be modeled by an interaction graph, in which edges represent pairs of nodes that attend to each other. Unlike message passing architectures, graph transformers have an interaction graph that is separate from the input graph. The typical interaction graph is a complete graph, which signifies a full attention mechanism that models direct interactions between all pairs of nodes. However, this creates quadratic computational and memory bottlenecks that limit the applicability of graph transformers to datasets on small graphs with at most a few thousand nodes. Making graph transformers scalable has been considered one of the most important research directions in the field (see the first open problem here).
A natural remedy is to use a sparse interaction graph with fewer edges. Many sparse and efficient transformers have been proposed to eliminate the quadratic bottleneck for sequences, however, they do not generally extend to graphs in a principled manner.
In “Exphormer: Sparse Transformers for Graphs”, presented at ICML 2023, we address the scalability challenge by introducing a sparse attention framework for transformers that is designed specifically for graph data. The Exphormer framework makes use of expander graphs, a powerful tool from spectral graph theory, and is able to achieve strong empirical results on a wide variety of datasets. Our implementation of Exphormer is now available on GitHub.
Expander graphs
A key idea at the heart of Exphormer is the use of expander graphs, which are sparse yet well-connected graphs that have some useful properties — 1) the matrix representation of the graphs have similar linear-algebraic properties as a complete graph, and 2) they exhibit rapid mixing of random walks, i.e., a small number of steps in a random walk from any starting node is enough to ensure convergence to a “stable” distribution on the nodes of the graph. Expanders have found applications to diverse areas, such as algorithms, pseudorandomness, complexity theory, and error-correcting codes.
A common class of expander graphs are d-regular expanders, in which there are d edges from every node (i.e., every node has degree d). The quality of an expander graph is measured by its spectral gap, an algebraic property of its adjacency matrix (a matrix representation of the graph in which rows and columns are indexed by nodes and entries indicate whether pairs of nodes are connected by an edge). Those that maximize the spectral gap are known as Ramanujan graphs — they achieve a gap of d - 2*√(d-1), which is essentially the best possible among d-regular graphs. A number of deterministic and randomized constructions of Ramanujan graphs have been proposed over the years for various values of d. We use a randomized expander construction of Friedman, which produces near-Ramanujan graphs.
 |
| Expander graphs are at the heart of Exphormer. A good expander is sparse yet exhibits rapid mixing of random walks, making its global connectivity suitable for an interaction graph in a graph transformer model. |
Exphormer replaces the dense, fully-connected interaction graph of a standard Transformer with edges of a sparse d-regular expander graph. Intuitively, the spectral approximation and mixing properties of an expander graph allow distant nodes to communicate with each other after one stacks multiple attention layers in a graph transformer architecture, even though the nodes may not attend to each other directly. Furthermore, by ensuring that d is constant (independent of the size of the number of nodes), we obtain a linear number of edges in the resulting interaction graph.
Exphormer: Constructing a sparse interaction graph
Exphormer combines expander edges with the input graph and virtual nodes. More specifically, the sparse attention mechanism of Exphormer builds an interaction graph consisting of three types of edges:
- Edges from the input graph (local attention)
- Edges from a constant-degree expander graph (expander attention)
- Edges from every node to a small set of virtual nodes (global attention)
 |
| Exphormer builds an interaction graph by combining three types of edges. The resulting graph has good connectivity properties and retains the inductive bias of the input dataset graph while still remaining sparse. |
Each component serves a specific purpose: the edges from the input graph retain the inductive bias from the input graph structure (which typically gets lost in a fully-connected attention module). Meanwhile, expander edges allow good global connectivity and random walk mixing properties (which spectrally approximate the complete graph with far fewer edges). Finally, virtual nodes serve as global “memory sinks” that can directly communicate with every node. While this results in additional edges from each virtual node equal to the number of nodes in the input graph, the resulting graph is still sparse. The degree of the expander graph and the number of virtual nodes are hyperparameters to tune for improving the quality metrics.
Furthermore, since we use an expander graph of constant degree and a small constant number of virtual nodes for the global attention, the resulting sparse attention mechanism is linear in the size of the original input graph, i.e., it models a number of direct interactions on the order of the total number of nodes and edges.
We additionally show that Exphormer is as expressive as the dense transformer and obeys universal approximation properties. In particular, when the sparse attention graph of Exphormer is augmented with self loops (edges connecting a node to itself), it can universally approximate continuous functions [1, 2].
Relation to sparse Transformers for sequences
It is interesting to compare Exphormer to sparse attention methods for sequences. Perhaps the architecture most conceptually similar to our approach is BigBird, which builds an interaction graph by combining different components. BigBird also uses virtual nodes, but, unlike Exphormer, it uses window attention and random attention from an Erdős-Rényi random graph model for the remaining components.
Window attention in BigBird looks at the tokens surrounding a token in a sequence — the local neighborhood attention in Exphormer can be viewed as a generalization of window attention to graphs.
The Erdős-Rényi graph on n nodes, G(n, p), which connects every pair of nodes independently with probability p, also functions as an expander graph for suitably high p. However, a superlinear number of edges (Ω(n log n)) is needed to ensure that an Erdős-Rényi graph is connected, let alone a good expander. On the other hand, the expanders used in Exphormer have only a linear number of edges.
Experimental results
Earlier works have shown the use of full graph Transformer-based models on datasets with graphs of size up to 5,000 nodes. To evaluate the performance of Exphormer, we build upon the celebrated GraphGPS framework [3], which combines both message passing and graph transformers and achieves state-of-the-art performance on a number of datasets. We show that replacing dense attention with Exphormer for the graph attention component in the GraphGPS framework allows one to achieve models with comparable or better performance, often with fewer trainable parameters.
Furthermore, Exphormer notably allows graph transformer architectures to scale well beyond the usual graph size limits mentioned above. Exphormer can scale up to datasets of 10,000+ node graphs, such as the Coauthor dataset, and even beyond to larger graphs such as the well-known ogbn-arxiv dataset, a citation network, which consists of 170K nodes and 1.1 million edges.
 |
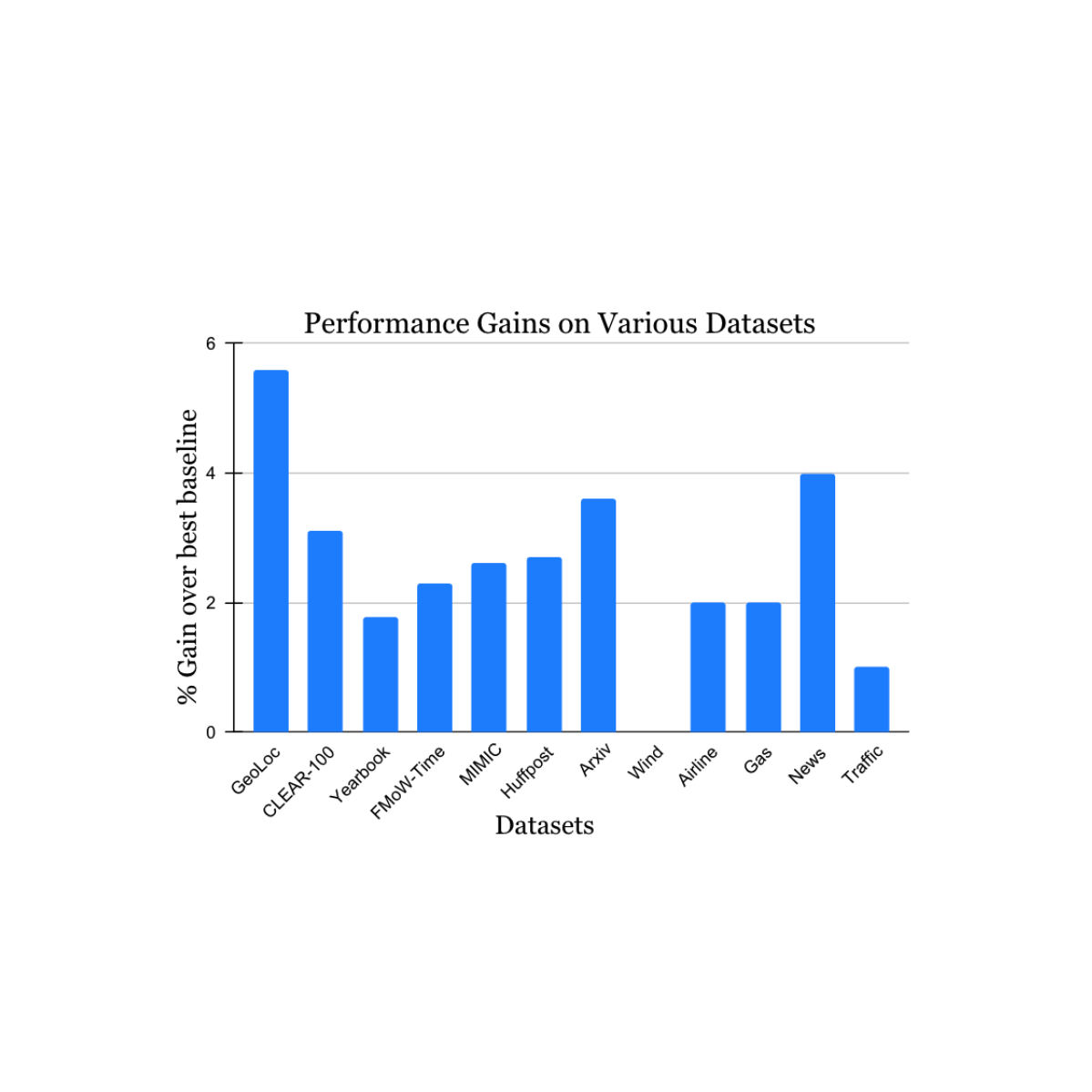
| Results comparing Exphormer to standard GraphGPS on the five Long Range Graph Benchmark datasets. We note that Exphormer achieved state-of-the-art results on four of the five datasets (PascalVOC-SP, COCO-SP, Peptides-Struct, PCQM-Contact) at the time of the paper’s publication. |
Finally, we observe that Exphormer, which creates an overlay graph of small diameter via expanders, exhibits the ability to effectively learn long-range dependencies. The Long Range Graph Benchmark is a suite of five graph learning datasets designed to measure the ability of models to capture long-range interactions. Results show that Exphormer-based models outperform standard GraphGPS models (which were previously state-of-the-art on four out of five datasets at the time of publication).
Conclusion
Graph transformers have emerged as an important architecture for ML that adapts the highly successful sequence-based transformers used in NLP to graph-structured data. Scalability has, however, proven to be a major challenge in enabling the use of graph transformers on datasets with large graphs. In this post, we have presented Exphormer, a sparse attention framework that uses expander graphs to improve scalability of graph transformers. Exphormer is shown to have important theoretical properties and exhibit strong empirical performance, particularly on datasets where it is crucial to learn long range dependencies. For more information, we point the reader to a short presentation video from ICML 2023.
Acknowledgements
We thank our research collaborators Hamed Shirzad and Danica J. Sutherland from The University of British Columbia as well as Ali Kemal Sinop from Google Research. Special thanks to Tom Small for creating the animation used in this post.